
In the highly regulated and competitive world of drug development, clinical trial protocol development is crucial for the success of clinical studies. A protocol is not just a set of instructions; it is in fact, a strategic document that shapes how a clinical study is conducted from start to finish. A poorly developed protocol can delay approvals, inflate budgets, and jeopardize data integrity. However, a well-crafted one, ensures clarity and alignment with all stakeholders, optimizes timelines, and increases the likelihood of regulatory and commercial success.
As a full-service CRO with deep experience across Central and Eastern Europe and beyond, Comac Medical understands the risks of missteps and the rewards of thoughtful protocol design. This article shares a practical, experience-based guide to developing a clinical trial protocol that is scientifically robust, operationally feasible, and aligned with global standards.
1. The Critical Role of Clinical Research Study Protocol Development
The clinical trial protocol definition is straightforward: it is the official document that describes the objectives, design, methodology, statistical considerations, and operational aspects of a clinical research study. It is arguably the single most important document governing trial conduct. Much like a blueprint guides the construction of a house from foundation to finish, the protocol guides the clinical trial from first patient in to final data lock, ensuring every component is aligned, compliant, and purposeful.
Regulatory bodies such as the FDA and EMA evaluate a protocol not only for its scientific accuracy but for its clarity, feasibility, and adherence to ethical and safety standards. International standards like ICH E6(R3) Good Clinical Practice (GCP), the SPIRIT guidelines for interventional trials, and the CONSORT standards for reporting all offer frameworks that shape the clinical trial protocol format.
Moreover, the protocol development process impacts every trial outcome including but not limited to the timelines, patient recruitment, cost, compliance, and data quality too. Clinical trial protocol amendments are not only time-consuming but also significantly increase development costs. According to a study by the Tufts Center for the Study of Drug Development (CSDD), biopharmaceutical companies implement an average of three amendments per Phase I protocol and up to seven amendments for Phase II and III protocols. The cost to implement each amendment ranges from approximately $250,000 to $450,000 in the US, not including the additional delays in timelines and resource reallocation. These figures highlight the critical importance of developing a robust, well-designed protocol from the outset to minimize costly revisions later in the trial lifecycle.
These delays typically reflect preventable study design flaws: overly complex procedures, poorly defined eligibility criteria, unrealistic scheduling, and lack of stakeholder input. That’s why clinical trial protocol optimization is more than good practice, it is arguably, a competitive advantage.
2. Top 5 Best Practices in Clinical Trial Protocol Development
To help sponsors reduce risk and build high-performing studies, here are five proven best practices based on real-world protocol experience.
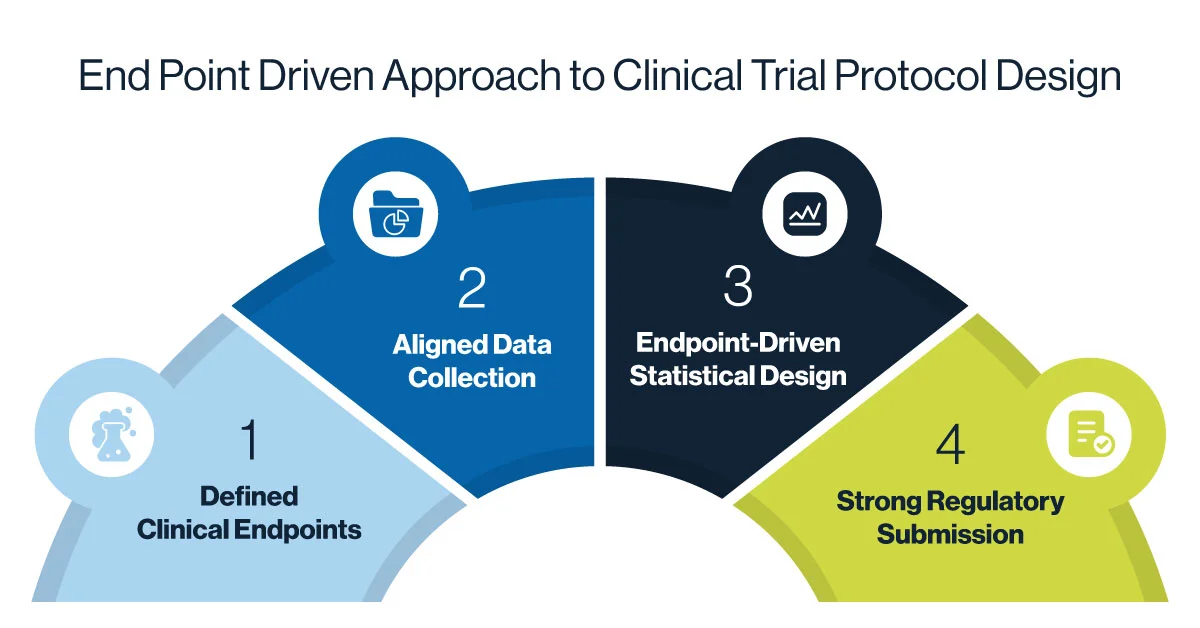
2.1 Embrace an End-point driven Approach
Every protocol should begin with a clear understanding of its endpoints. Specifically, what does clinical trial success look like, both clinically and statistically? Determination of primary and secondary endpoints are essential to decide before beginning the trial. In effective clinical trial protocol design, endpoints shape everything – from inclusion and exclusion criteria to visit schedules, assessments, and statistical analysis.
For example, if the primary endpoint is a 12-week improvement in a dermatologic condition, data collection must align closely with that timeframe. The sample size and power calculation must be anchored to the expected effect size. Without this alignment, a study may collect vast amounts of irrelevant data, miss its endpoint window, or be underpowered for meaningful conclusions.
As the FDA notes in its guidance on adaptive design, a well-defined primary endpoint and pre-specified analysis plan reduce the risk of bias and support regulatory confidence in trial results.
2.2 Involve Stakeholders Early
Successful clinical trial protocol development is never conducted in isolation. Protocols that involve only medical writing or clinical staff often miss key operational, regulatory, and statistical insights. Early engagement with all functional departments including clinical operations, data management, biostatistics, pharmacovigilance, clinical pharmacology and regulatory affairs. This interdisciplinary coordination ensures that the protocol is both scientifically accurate and operationally viable and may reduce the likelihood for amendments.
It is also important to include patient and investigator feedback. Patient-centric protocols support better adherence and recruitment. Investigator input can flag burdensome elements or identify better practices for assessments and monitoring.
2.3 Build Flexibility into the Clinical Protocol Development Design
Protocol amendments are not always avoidable, but they can often be minimized by building in appropriate flexibility. That means allowing reasonable visit windows, offering optional assessments, specifying dosage ranges, and defining stopping rules in advance. This kind of adaptive thinking is increasingly encouraged by regulators and can be a valuable differentiator for emerging biotech companies.
Flexible clinical trial protocol formats also support faster start-up across multiple geographies and make it easier to adjust based on early site or patient feedback without compromising scientific integrity. In line with the new ICH E6(R3) Guidelines for Good Clinical Practice, applying Quality by Design (QbD) principles during clinical trial protocol development ensures potential risks and critical factors are anticipated and mitigated early. Pre-defined thresholds (i.e. quality tolerance limits, QTLs) set before clinical trial protocol finalization make it easier to identify potential issues that could affect the reliability of trial results and could improve trial efficiency and reduce unnecessary delays caused by unexpected deviations.
2.4 Document the Rationale Throughout
Regulatory bodies are not just reviewing what you propose, they’re asking why. Documenting the rationale for each major decision (e.g., why a certain inclusion criterion was chosen or why a biomarker is used) strengthens the protocol and prepares you for submission, inspection, or future adaptations.
Clear rationale also reduces internal confusion, accelerates alignment between sponsors and CROs, and supports knowledge transfer when teams change. If you’re using a clinical trial protocol template, ensure it includes rationale sections or embedded commentary that can be edited or hidden as needed.
2.5 Prioritize Real-World Feasibility
Even the most scientifically rigorous protocol can fall short if it isn’t feasible to execute in real-world settings. A protocol that overburdens patients or exceeds site capabilities risks poor recruitment, high deviation rates, and delays in study timelines. Feasibility must be built into the design phase, not retrofitted later. This means aligning procedures with standard clinical practice, minimizing unnecessary assessments, and considering geographic differences across trial sites.
Engaging experienced primary investigators, site staff, and clinical operations teams early can help flag potential obstacles before they become costly issues. Real-world input ensures the protocol is not only scientifically sound, but also practical, executable, and patient-friendly, an increasingly critical factor for study success and regulatory confidence.

3. Top 5 Common Pitfalls to Avoid in Protocol Development
Despite the availability of global clinical trial protocol guidelines, many common pitfalls persist in protocol writing. These mistakes cost sponsors both time and credibility.
3.1 Overcomplexity: When More Becomes Less
One of the most common and costly mistakes in clinical trial protocol development is making the protocol too complex. Sponsors often add multiple exploratory endpoints, excessive visit procedures, or redundant data points in an attempt to “future-proof” the trial or satisfy multiple internal stakeholders. However, this approach backfires. Overcomplex protocols are harder to implement for sites, slow to recruit, and more prone to deviations.
How to prevent it:
Focus on your primary and key secondary endpoints. Every procedure or assessment should map directly to those endpoints. Use cross-functional protocol review to challenge each element: Is it necessary? Is it feasible? Will it yield actionable data? Prioritize clarity and efficiency over comprehensiveness.
3.2 Vague Eligibility Criteria
Protocols that include vague, inconsistent, or poorly defined language create confusion for investigators and site staff. Ambiguity in inclusion/exclusion criteria, visit schedules, or procedural steps can result in inconsistent patient screening and data collection, and lead to increased protocol deviations or audit findings.
How to prevent it:
Use precise, operational language and avoid subjective terms unless they are clearly defined. Also, ensure protocol reviewed by operational teams, including site-facing personnel, to ensure instructions are interpretable and unambiguous in practice.
3.3 No Country-level Adaptation
A protocol that looks solid on paper may fail when implemented across different countries and health systems. Therefore, ignoring local standards of care, regulatory requirements, or cultural and logistical realities can hinder start-up timelines and slow down patient enrollment.
How to prevent it:
Engage a CRO or feasibility expert early in the clinical trial protocol development process to provide region-specific insights. For example, in Central and Eastern Europe, access to certain diagnostics or treatment modalities may differ from Western Europe or the US. Local principal investigator input is extremely valuable in identifying protocol design elements that may be impractical or misaligned with clinical practice in each region.
3.4 High Patient Burden
Even if a protocol is scientifically sound and operationally feasible, it may still fail if it doesn’t consider the patient’s perspective. Complex visit schedules, invasive procedures, or long travel times deter enrollment and increase dropout rates, especially in outpatient or chronic disease studies.
How to prevent it:
Design with the patient in mind. Include patient advisors during protocol development where possible. Simplify visit structures, use patient-distractive environment during some trial procedures when applicable (e.g. music, television), use remote assessments when feasible, and consider the cumulative burden on participants over the study duration. A patient-friendly protocol is not just ethical, it’s more likely to succeed.
3.5 Skipped Internal Reviews
In fast-moving environments, sponsors sometimes push protocols through internal review with insufficient input from key functions or limited time for meaningful revisions. This leads to avoidable design errors, misaligned expectations, and gaps that surface only after submission or study initiation.
How to prevent it:
Establish a structured, cross-functional protocol review process with realistic timelines. Ensure all involved departments such as clinical operations, biostatistics, medical, regulatory, and quality assurance all can review and provide feedback. Track and document major decisions and changes, this not only improves protocol quality but also strengthens your regulatory submission and audit readiness.

4. How a CRO Like Comac Medical Adds Value
Biotech sponsors often face tight timelines and limited internal bandwidth. This makes the choice of CRO partner especially critical during clinical trial protocol development.
At Comac Medical, we support protocol success from the earliest stages. Our experience spans over 38 countries, with deep specialization in Central and Eastern Europe. We offer:
✔️ Real-world feasibility insights from our network of investigators and sites
✔️ Protocol localization expertise for regional adaptation
✔️ Cross-functional protocol advisory (clinical, regulatory, medical writing, and biostats)
✔️ Rapid yet rigorous internal reviews to catch design gaps early
✔️ A track record of reducing the number and severity of amendments across therapeutic areas
By involving our team during protocol drafting, not just after, we help sponsors avoid costly missteps and ensure their protocols are both compliant and executable.
Read more about Comac Medical’s services here.
5. A Strong Protocol Is a Strategic Advantage
The importance of foresight, clarity, and collaboration cannot be overstated. A well-designed protocol reduces downstream risk, builds regulatory trust, and sets the entire study on a path to success.
Biotech sponsors who treat protocol design as a strategic investment, not a paperwork task, gain an edge in both operational efficiency and data quality. And with the support of an experienced, regionally aware CRO like Comac Medical, your protocol can become the catalyst for a high-performing trial, not a hurdle to overcome.


